|
||||||||||||
DNA Analysis RulesThe following rules summarize an extensive project to standardize the way DNA analysis by flow cytometry should be performed in order to minimize differences between individuals interpreting the data. Rules for Obtaining High Quality DNA Histograms and Optimizing Correlation of S-phase Estimates Between OperatorsNote: This section may change over time as additional data is obtained from participating laboratories. I. Acquisition RulesA. ParameterThe DNA fluorescence parameter should be either a linear integral or area type of signal. It is important to maintain a consistent gain setting. See further under point F in Acquisition Rules. B. LinearityThe linearity of the DNA fluorescence amplifier should be tested. Non-linear amplifiers should not be used for DNA analysis. C. DiscriminatorEvents should be discriminated on the DNA fluorescence parameter only (e.g. red fluorescence for Propidium Iodide). The discriminator level should be as low as possible without creating a debris peak that is greater than the highest G0G1 peak. D. GatingGating should not be performed during acquisition. Gating is only recommended for multiparameter DNA histograms such as cytokeratin vs DNA or BrdUrd vs DNA. Gating on light scatter is not recommended due to the heterogeneity of the distribution. Signal processing gating, signal peak height vs signal peak area, to eliminate aggregates is only recommended if the aggregates are clearly and completely separated from singlet particles which is usually only true for experimental tissue culture cell lines. E. Number of Events and ResolutionSimulation studies indicate that for accurate S-phase estimates, there should be an average of approximately 100 events per channel between the lowest G1 and highest G2 of the histogram when the resolution is 256 channels. If a histogram has its diploid G0G1 on channel 50 and the last G2 of an aneuploid population is at 200, there should be at least 15,000 events between channels 50 and 200. F. Location of diploid G0G1The position of the DNA Diploid G0G1 peak should always be placed in about the same channel. For 256 channel histograms, the recommended location is channel 50. For 1024 channel histograms (not normally recommended because it requires collection of 400% more events for same histogram at 256 channels), the location is channel 200. G. Changing Gains1. Normally adjust the gain to center the DNA diploid peak on a particular channel (e.g. 50). 2. When a hyper-tetraploid population is observed during acquisition, it is desirable to reduce the gain so that its G2M and some channels consisting of only background are on scale. 3. Note, after adjusting gain, acquisition must be restarted. Gain should be reset to the normal location when running the next sample. H. Time or Chronology ParameterIf the instrument supports either time or chronology, it is highly recommended to view a time vs. DNA parameter to detect any peak shifts during acquisition.
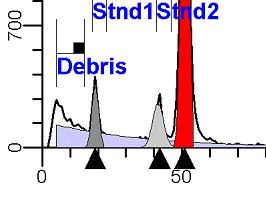
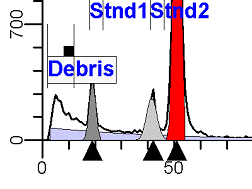
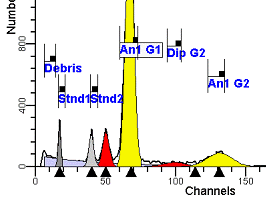
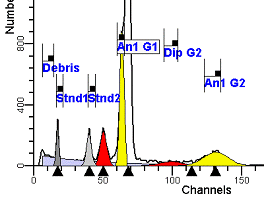
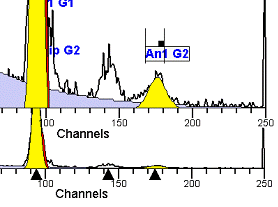
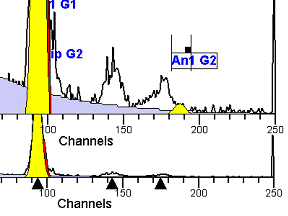
II. Analysis RulesA. General Procedure1. Analyze all histograms in automatic analysis mode. a) For best results, try to use settings for the program’s configuration, peak finding characteristics and automatic analysis properties that results in most histograms being analyzed accurately. 2. Evaluate and review each stored analysis report and re-analyze if the model used is incorrect for the data. 3. If available, run automatic linearity adjustment on all reports. 4. Final review. B. Reviewing Process1. Model Selection Check The most important step in analyzing DNA histograms in a consistent manner is checking the correct ploidy model for a particular DNA histogram. In some cases this process may require several analyses to achieve the correct and optimal fit, i.e. the RCS value should be as low as possible (< 3.0). Use the rules below to help guide you through this process. a) General Considerations (1) If two model components are of similar shape and are highly overlapped (>75%), it may be necessary to add additional constraints to the model or, in the worse case, disable the model component of lesser importance. (2) Always make the G2M position and standard deviation dependent on its associated G1 by some linearity factor unless, (a) The cell cycle analysis program cannot find the optimal linearity factor or (b) The histogram is highly non-linear where there is either compression or contraction of the scale at higher channel numbers. (3) Always model S-phase as a single, broadened rectangle. (4) When choosing between two very similar models, select the one that gives consistent results with slightly different range settings. (a) An example of this rule might be when trying to use an aneuploid model with a near-tetraploid type of histogram. If the aneuploid model only works with very specific range settings, choose the tetraploid model instead. b) Tetraploid Model Selection (1) Select a tetraploid model if the DI is close to the expected diploid G2/G1 ratio, (a) Use +/- 0.15 ratio units as a guide but note that if the diploid G2 is not modeled properly, a tetraploid model may be necessary even though it falls outside of the above range. (2) and there is another peak at (8C) that cannot be explained as an aggregate. (a) Consider the 8C peak to be an aggregate if it is less than the 6C peak. (3) Choose a tetraploid model over an aneuploid model if the diploid G2M overlaps too significantly with an aneuploid G1. (a) The diploid G2M will generally only model properly if there is a clearly distinguished peak at its expected location. (b) Inappropriate fits of the diploid G2M are usually associated with a zero or very high calculated percentage or a location that results in a G2/G1 ratio outside the expected range. c) Aneuploid Model Selection (1) Only choose this model if the potential aneuploid’s G0G1 cannot be explained as an aggregate or some other part of another cycle (e.g. G2M). (2) and there are adequate channels to model the entire cycle. d) Near-diploid Model Selection (1) Choose the near-diploid model if the two G0G1 peaks can be clearly distinguished and the resulting fit seems appropriate. (2) If the DI is between 0.7-1.0 or 1.0-1.3 disable the diploid S-phase and make both G2’s dependent. (3) For very near-diploids, it may be necessary to force the standard deviations of the two G0G1 model components to be equal to yield an appropriate fit. e) Hypo-diploid Model Selection (1) Only select a hypo-diploid model when there are standards or normal controls that accurately determine the expected diploid G0G1 position. (2) If the hypo-diploid G0G1 overlaps one of the standards, disable the standard model component and re-model. (3) Near-diploid rules apply. 2. Range Positions Check The most common reason for uncorrelated results between two fits using the same model is inattention to range positions. Do not change a range setting unless it is necessary to do so. a) Debris Range (1) The beginning of the debris range should correspond to the channel with the highest debris counts (see Figure 1, Range: Debris for examples of correct and incorrect placements). b) Peak Ranges 1) Center range about the peak and make sure estimates appropriately fit the data (see Figure 1, Range: Peak G0G1 and Range: Peak G2M examples of correct and incorrect placements). (2) Exceptions to centering the range are for near-diploid and near-tetraploid G0G1 peaks. These ranges need to be displaced to yield reasonable estimates for the underlying peaks. Figure 1
Proficiency RequirementsQuestion: Answer: Since our intent was to demonstrate that this test could be standardized, we also had to tackle the detail of how to eliminate operator variability and bias in DNA histogram analysis. In other words, it was not good enough for one person to analyze all the DNA histograms and show the highly significant prognostic categories. We had to show that anyone, if trained properly, could obtain equivalent results. This problem was initially intractable since there were so many areas in the modeling decision-making that resulted in similar results. It wasn't until we changed the problem from obtaining similar results to identical results that we started making good headway solving this problem. The process started with two operators independently analyzing a single DNA histogram. If there were any difference at all, we hunted down the reasons behind this difference and created rules that if followed would eliminate the difference. In other words, by tolerating no difference (<0.01% S-phase difference) we were able to start creating a concrete rule set. This process took us far along to an eventual solution, but not all the way. There were still some model decisions that could not be eliminated by simple rules. One was the input of a linearity factor and the other involved the relationship between the G2M and G1 peaks. Within any decent modeling program there is a factor that is used to control the aggregate model component and the relationship between G2M and G1, if they happen to be dependent on each other multiplicatively. The user has traditionally entered this linearity factor. We found there were biases in its selection, which ultimately changed the results. We finally eliminated this source of error by creating an AutoLinearity adjustment in ModFitLT 3.1 that automatically finds the best linearity factor for a given DNA histogram. The other intractable problem we had was the determination of whether a G2M should be dependent on G1 or should be allowed to float. Traditionally, this decision was based on a rather subjective decision of whether the G2M was clearly visible or not. This decision was confounded by the fact that other neighboring model components such as an aggregate doublet or another ploidy population could destabilize the position of the G2M. We initially tried to fix this problem by creating criteria rules regarding the position of the G2M, but they kept getting more complicated and were impossible to standardize between operators. We finally solved this problem by making all default models in ModFitLT 3.1 and above use dependent G2M's. In other words, the position of the G2M's is dependent on the position of the G1 and the calculated optimal linearity factor. This approach resulted in operators obtaining nearly identical results (<0.1% difference in S-phase). We've successfully used this training method in many flow cytometry courses without much difficulty. Given all this discussion, here is our answer. If a prospective operator takes the proficiency test and there are any red marks on any of the pages, they must go through the training of the relevant sections again. A red score is made if any S-phase result is off by 0.1%. Even though this criteria is far less than what is clinically significant, it is still logical to hold the prospective operator to this high standard. The reason is that if they are off by 0.1% or more, they did not follow one or more of the rules and are likely to not follow them for future clinical samples or they are using the wrong version of ModFitLT (version <3.1). It may be that no matter how many times they go through the training, they just can't get a few files. Under this condition they should go back to our reference reports and understand how we analyzed the files differently. It's possible that they will still disagree with our analysis. Under that condition they can challenge the analysis. Initially, Verity will need to respond to that challenge, but eventually someone there at your laboratory can do it as well. There are three results possible from a challenge(s): 1) they didn't see one of the rules in operation and will learn from the experience, 2) we made a mistake and didn't follow one of our rules or 3) the rules result in an ambiguous situation resulting in two or more correct approaches. The first possibility is just part of the learning process. The second possibility would be corrected immediately by Verity and their analysis stands. The third possibility might necessitate a rule change for the next version of the training system. We believe the above operating procedure is the only logical way to proceed. It sounds initially like it can't work, but we think it can. It sounds like it might be too expensive in technician time, but we think the reverse will be true. Creating a really tough proficiency exam at the outset will have long-term benefits that will outweigh the multiple passes through the training system. There are likely to be problems with already "DNA histogram analysis savy" technicians. We must expect this problem and attribute it to a process of breaking down biases. |
||||||||||||
Your Cart Is Empty